reverse-zMAP module
Functional Description:
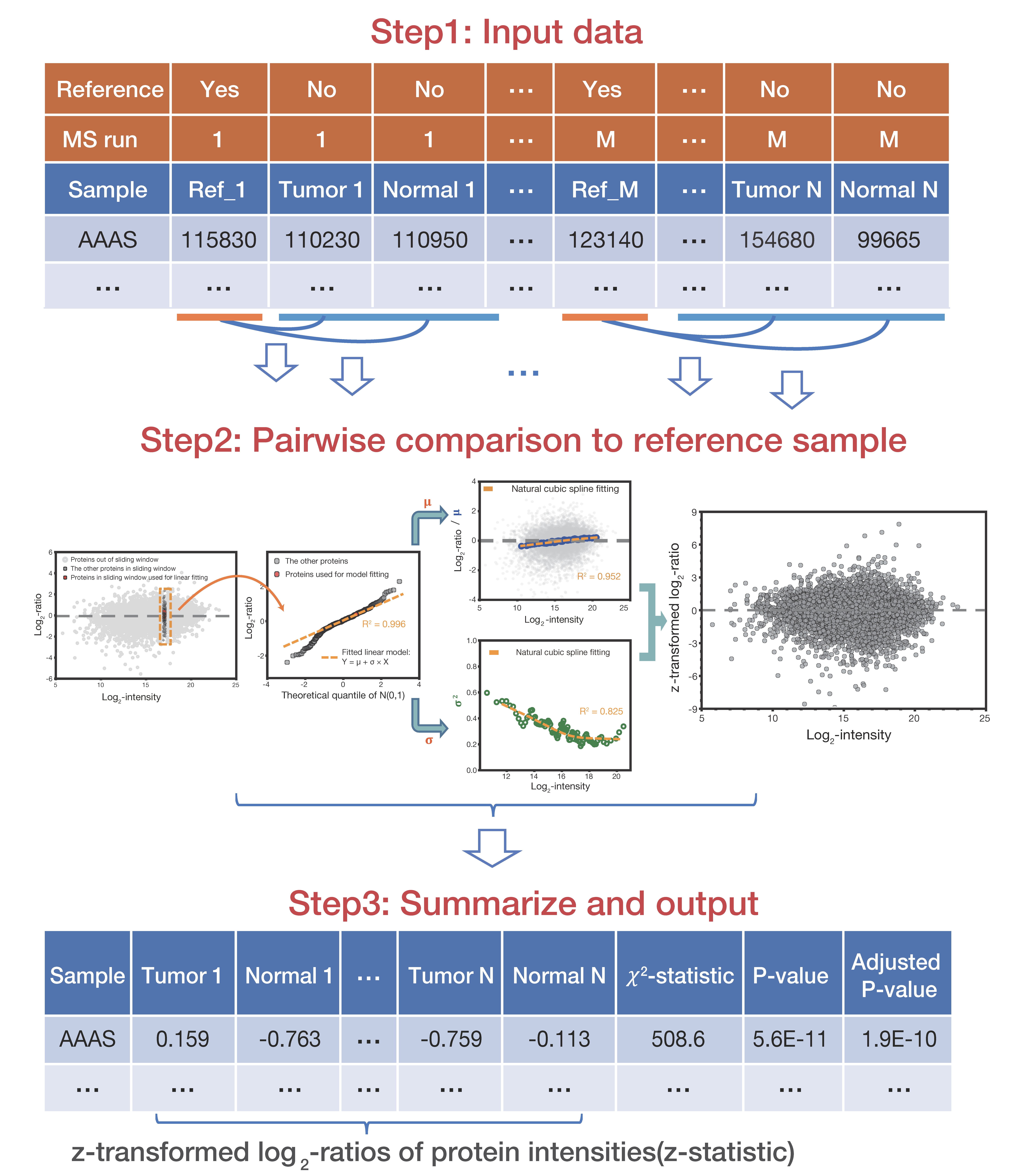
reverse-zMAP module is primarily designed for handling MS runs with relatively large sample sizes.
The major concern is that, with the increase of the number of samples, the odds that outlier measurements
are involved for each specific protein increase, giving rise to excessively large variances. Consequently,
in the sliding-window process of the zMAP module, the proportion of proteins that are suitable to use for the
quantile regression in each window can be very small. Besides, fitting a single mean-variance curve (MVC) for a
large number of samples may not be flexible enough to allow for the variation of mean-variance trend across samples.
In practice, large-scale proteomic studies have frequently applied the strategy of adding a biologically identical
reference sample to each individual MS run. For example, in cancer studies, a mixed sample is typically generated
by pooling tumor samples and/or normal adjacent tissues (NATs) from several related patients in equal protein amounts.
The proteome of this mixed sample is then profiled in every MS run separately. reverse-zMAP module alleviates the influence
of outliers by repeatedly making pairwise comparisons, which in the meanwhile allows the modeling of sample-specific
mean-variance trend, but it requires a biologically identical reference sample in each MS run for a subsequent integration across MS runs.
A test example is:
Protein intensity file(tab-delimited): reverse_zMAP_example_protein_intensity.txt
Sample information file(tab-delimited): reverse_zMAP_example_sample_info.txt